



The cardiovascular system comprises the heart, veins, arteries, and capillary beds. The atrioventricular (mitral and tricuspid) and semilunar (aortic and pulmonic) valves keep blood flowing in one direction through the heart, and valves in large veins keep blood flowing back toward the heart. The rate and force of contraction of the heart and the degree of constriction or dilatation of blood vessels are determined by the autonomic nervous system (sympathetic and parasympathetic) and hormones produced either by the heart and blood vessels (ie, paracrine or autocrine) or at a distance from the heart and blood vessels (ie, endocrine).
Heart, cow
Healthy bovine heart. Illustration by Dr. Gheorghe Constantinescu. |

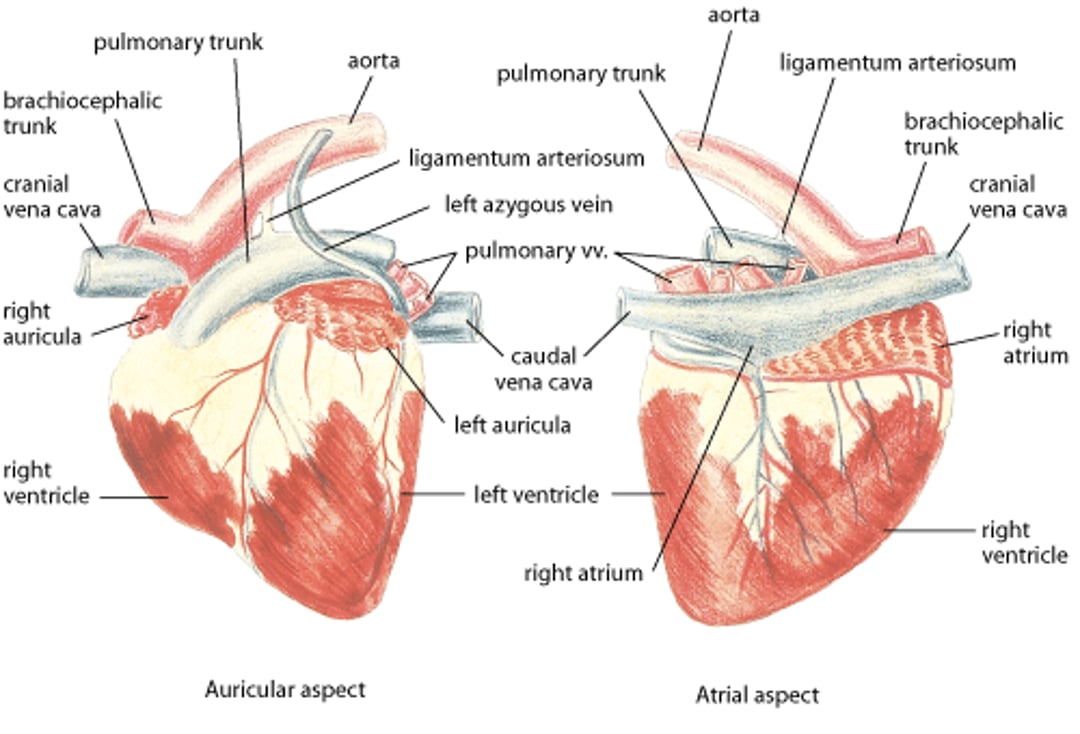
Slightly >10% of all domestic animals examined by a veterinarian have some form of cardiovascular disease (clinically significant or insignificant), with varying prevalences of cardiac disease based on species, breed, and etiology (congenital vs acquired cardiovascular disease). The true prevalence of cardiovascular disease is likely underestimated because the majority of domestic animals do not receive a cardiac workup.
Similar to many chronic diseases of other organ systems, cardiovascular diseases generally do not resolve but progress and become more limiting over time, which may ultimately lead to death. Evaluation of the heart is performed via assessment of:
heart sounds and murmurs
arterial pulses
degree of jugular vein distention
strength and location of the apex beat
electrocardiography
radiography
cardiac biomarkers
echocardiography
other advanced imaging techniques, such as
angiography
fluoroscopy
CT scan with angiography
cardiac MRI
Heart Rate and the Electrocardiogram

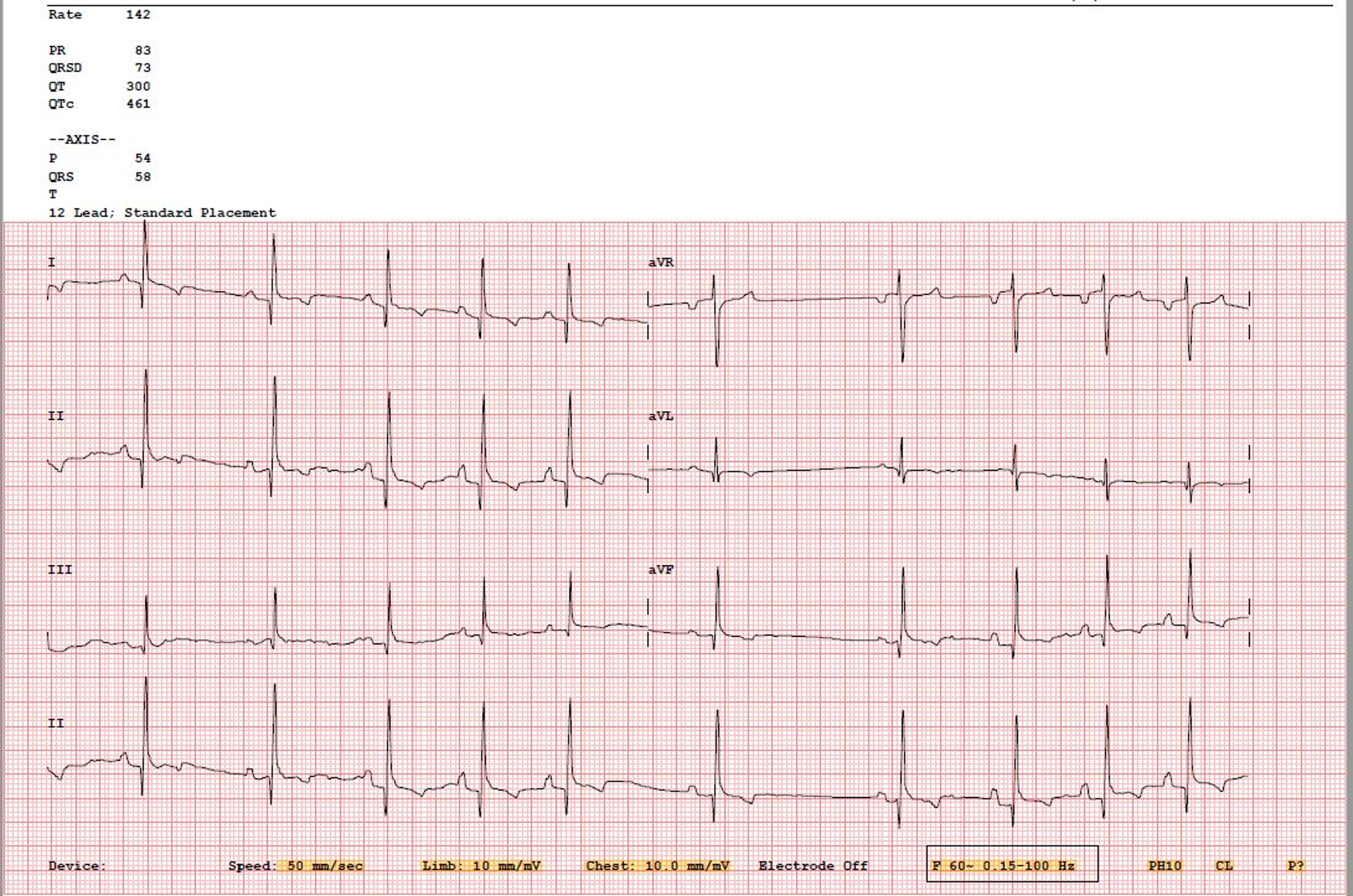
Courtesy of Dr. Kursten V. Pierce.

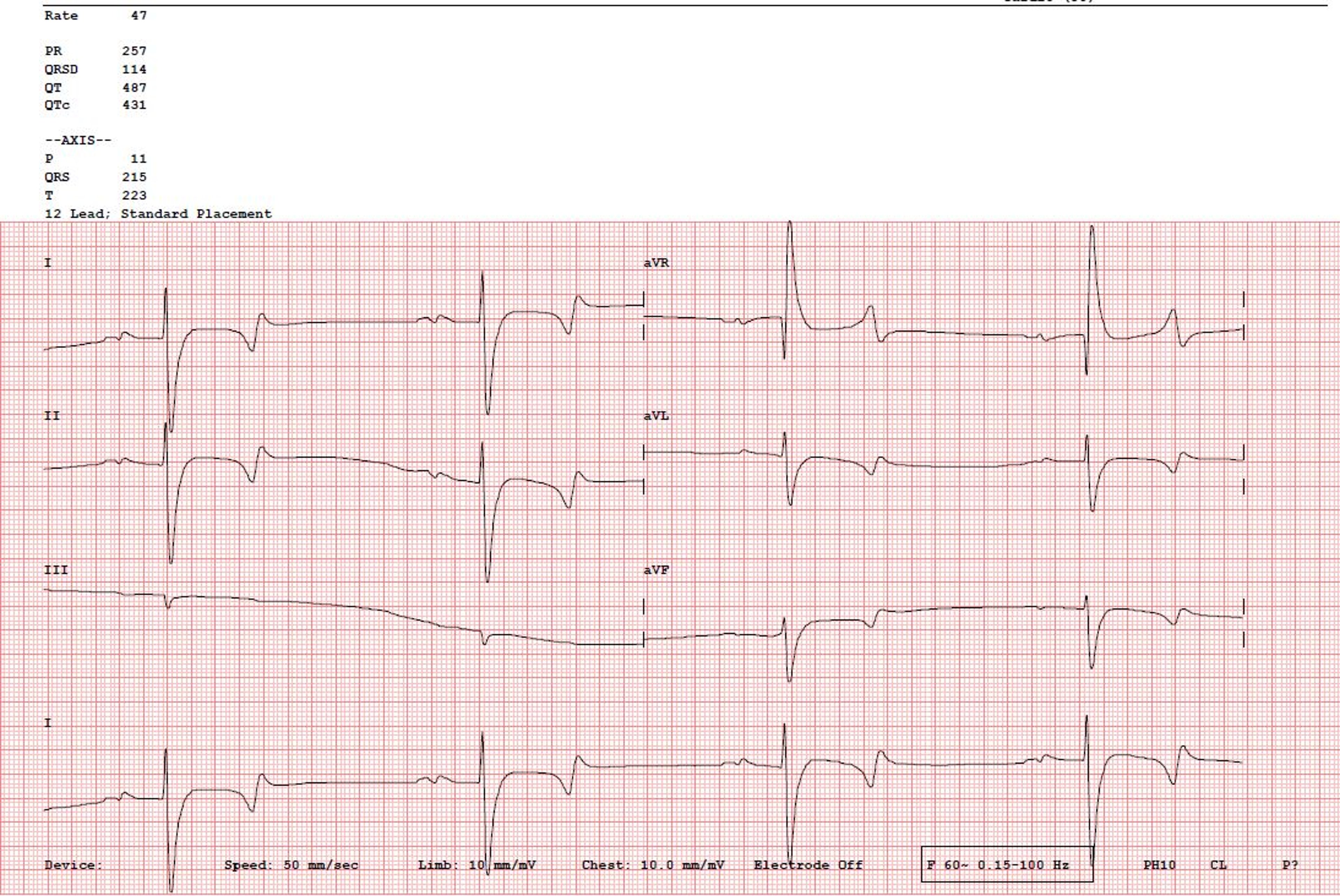
Courtesy of Dr. Kursten V. Pierce.
The sinoatrial (SA) node is the pacemaker of the heart. The heartbeat originates with a wave of depolarization that begins in the SA node at the juncture of the cranial vena cava and the right atrium. At rest, the SA node discharges ~30 times/min in horses, >120 times/min in cats (typically 180–220 times/min in a hospital setting), and 60–120 times/min in dogs (range of 40–260 bpm, with average daily rate of 80 bpm for an adult dog based on 24-hour Holter monitoring), depending on their size. In general, the larger the species, the slower the rate of SA node discharge and the slower the heart rate. Birds can have a resting heart rate of ~115–130 beats/min, with active heart rates up to 670 beats/min, depending on size and species. Hummingbirds can have an active heart rate of >1,200 beats/min.
The rate of SA nodal discharge increases when norepinephrine (released from the sympathetic nerves) or epinephrine (released from the adrenal medulla) binds to the beta1-adrenoreceptors on the SA node (sympathetic stimulation). This cardioacceleration (positive chronotropic effect) may be blocked by beta-adrenergic blocking agents (eg, propranolol, atenolol, metoprolol, esmolol, carvedilol). The rate of SA nodal discharge decreases when acetylcholine released by the parasympathetic (vagus) nerves binds to the cholinergic receptors on the SA node (vagal stimulation). This vagally mediated cardiodeceleration (negative chronotropic effect) may be blocked by a parasympatholytic (vagolytic) compound (eg, atropine, glycopyrrolate). When the SA node discharges and the wave of depolarization traverses the atria via preferential pathways (interatrial, internodal, and atrionodal) toward the atrioventricular (AV) node, the P wave (atriogram) of the ECG is produced. Subsequently, the atria contract, ejecting a small volume of remaining blood into the respective ventricles (atrial kick). Atrial repolarization (Ta) is typically difficult to visualize in small animals due to its low voltage deflection.
In quiet, healthy animals, the cyclic variation of the heart rate with respiration is termed respiratory sinus arrhythmia (RSA); it results from decreased vagal activity during inspiration (increase in SA node discharge rate with resultant increase in heart rate) and increased vagal activity during expiration (decrease in SA node discharge rate with resultant decrease in heart rate). In addition to variations in vagal tone with respiration, other mechanisms contribute to RSA, including response of the cardiopulmonary and baroreceptors (eg, Frank-Starling mechanism, Bainbridge reflex). Therefore, vagolytic compounds, as well as excitement, pain, fever, and congestive heart failure (CHF), usually abolish or diminish RSA. Heart rate variability synchronized with respirations is a good indicator of cardiac health. It is rare to find an animal that has active CHF with RSA; however, comorbid conditions that increase vagal activity (such as primary respiratory or neurologic disease) may cause RSA to persist. RSA is uncommonly documented in cats in the hospital setting due to their higher sympathetic tone.
Heart rate is also inversely related to systemic arterial blood pressure. When blood pressure increases, heart rate decreases; when blood pressure decreases, heart rate increases. This relation is known as Marey's Law and occurs by the following mechanism. When high-pressure arterial baroreceptors in the aortic and carotid sinuses detect increases in blood pressure, they send increased afferent volleys to the medulla oblongata, which increases vagal efferents to the SA node and causes the heart rate to decrease. In heart failure, the baroreceptors (laden with Na+/K+-ATPase) become fatigued, which reduces the afferent signals to the medulla oblongata. This results in less vagal efferent signaling. Thus, dogs in CHF have a decrease in heart rate variability and frequently present with an underlying sinus tachycardia.
Once the wave of depolarization reaches the atrioventricular (AV) node, the speed of conduction is slowed through the nodal tissue, giving the atria time to contract and eject more blood into the ventricles, allowing for atrioventricular synchrony. The depolarization then travels rapidly via a specialized conduction system (ie, bundle of His, right and left bundles branches, Purkinje network) to the subendocardium of the ventricles and to the ventricular septum. From these points, it travels slowly through the ventricular myocardium (subendocardial to epicardial in category A species), producing the QRS complex of the ECG (representing ventricular depolarization) with subsequent mechanical ventricular contraction.
The process of ventricular activation varies between domestic mammals, which are characterized as either category A (dog, human, monkey, cat, rat) or category B (goat, horse, cow, pig, sheep) based on the time and order of the 3 fronts (waves) of depolarization. Animals in category B have more extensive Purkinje fiber networks throughout the ventricular myocardium into the epicardium, thus the wave front does not spread from endocardium to epicardium as in category A mammals. For this reason, chamber enlargement cannot be determined from a base apex lead used for horses and large animals, and the ECG is only used to assess the cardiac rhythm. The delay between the electrical activity visualized on ECG and mechanical function accounts for transmission of impulses, which allows contraction of cardiomyocytes to occur in synchrony. Under rare conditions, there may be depolarization without contraction; this is called electromechanical dissociation.
The interval on an ECG between the onset of the P wave and the onset of the QRS complex is termed the PQ or PR interval. It is a measure of the time it takes for the electrical wave of depolarization to begin at the SA node, traverse the AV node, and reach the ventricles. Factors that speed or slow the rate of discharge of the SA node (chronotropy) also speed or slow conduction through the AV node (dromotropy). Thus, as the heart rate increases, the PR interval shortens; when heart rate slows, the PR interval lengthens.
The T wave of the ECG represents repolarization of the ventricles. It is affected by electrolyte imbalance (eg, hypo- or hyperkalemia, hypo- or hypercalcemia), myocardial injury, or ventricular enlargement. Repolarization of the atria (Ta wave) is rarely seen, because it occurs during the much larger QRS complex. Occasionally, it can be seen with AV nodal disease (AV block) or in horses with slow heart rates, appearing as a “hammock” after the P wave.
The electrocardiogram can be studied in three different anatomic planes, including frontal plane or X-axis of the Cartesian system, sagittal plane or Y-axis of the Cartesian system, and horizontal (transverse) plane or Z-axis of the Cartesian system. In addition, there are three main lead systems used in veterinary medicine for category A mammals: Bailey's hexaxial system (records electrical activity in frontal plane), precordial or chest lead system (records electrical activity in horizontal plane), and bipolar orthogonal system (records electrical activity in all three planes). In category B mammals, a base-apex lead configuration is used. Interpretation of ECGs should be performed in a systematic manner, including assessment of the heart rate and rhythm, evaluating waveforms (P, QRS, T) and segments (PR/PR, ST, QT), and assessing for presence of a P wave for every QRS wave and vice versa. An in-depth review of ECG interpretation is beyond the scope of this discussion.
Force of Ventricular Contraction
The force with which the ventricles contract is determined by many factors, including the end-diastolic volume (preload), which is the volume of blood within the ventricles just before they begin to contract, and myocardial contractility (inotropy), which is the rate of cycling of the microscopic contractile units of the myocardium.
The preload is determined by the difference in end-diastolic pressure between the ventricle and the pleural space, divided by the stiffness of the ventricular myocardium. The end-diastolic pressure of the ventricle is determined by the ratio of blood volume and the compliance of the myocardium. Preload is regulated predominantly by low-pressure volume receptors in the heart and large veins. When these receptors are stimulated by an increase in blood volume or by distention of the structures the receptors occupy, the body responds by making more urine and by dilating the veins—an attempt to decrease blood volume and lower the pressures in the veins responsible for venous distention. An increase in end-diastolic volume (preload) stretches the ventricular wall, resulting in a more forceful contraction as per the Frank-Starling mechanism, or Starling's law of the heart.
Stretching of receptors in the atria and ventricles causes them to release natriuretic proteins: brain natriuretic peptide (BNP) primarily from the ventricles and atrial natriuretic peptide (ANP) from the atria. These peptides are natriuretic, relax smooth muscle, and in general oppose vasopressin and angiotensin II. A correlation between BNP and NT-proBNP levels and degree of stretch of the heart has been identified in dogs.
Myocardial contractility is determined by the availability of ATP and calcium, which allows myosin-actin cross-bridging to occur. The rate of liberation of energy from ATP is determined, in part, by the amount of norepinephrine binding to beta1-adrenergic receptors in the myocardium. One of the most important factors in heart failure is the down-regulation (decreased number) of myocardial beta1-receptors.
Oxygen and the Myocardium
Oxygen is essential for the production of energy that permits all body functions. The amount of oxygen available for production of this energy is termed the tissue oxygen content. The myocardial oxygen content is a balance between how much oxygen is delivered to the heart minus how much oxygen is consumed by the heart.
The amount of oxygen delivered to the heart depends on how well the lungs function, how much Hgb is present to carry the oxygen, and how much blood carrying the Hgb flows through the heart muscle via the coronary arteries. If pulmonary function is normal and there is sufficient Hgb, coronary blood flow will determine how much oxygen is delivered to the myocardium. Coronary blood flow is determined by the difference in mean pressure between the aorta (normally 100 mm Hg) and the right atrium (normally 5 mm Hg), into which coronary blood empties. Because coronary flow is greatest during diastole, slower heart rates (which preferentially increase diastolic interval) are associated with improved myocardial oxygen delivery.
The amount of oxygen consumed by the heart is termed myocardial oxygen consumption. It is determined, principally, by wall tension and heart rate. Wall tension is expressed by the law of LaPlace, in which tension increases with increases in pressure or diameter of the ventricle, and tension decreases with increases in wall thickness of the ventricle. Tension increases with conditions that increase afterload (pressure), such as pulmonary valve stenosis, subaortic stenosis, systemic or pulmonary hypertension, or preload (volume), including mitral valve insufficiency, left-to-right shunting defects, and dilated cardiomyopathy. In the absence of a stenotic lesion, afterload is determined by the relative stiffness of the arteries and by the degree of constriction of the arterioles. The tone of vascular smooth muscle depends on many factors, some of which constrict the muscle (eg, adrenergic agonists, angiotensin II, vasopressin, endothelin) and some of which relax the muscle (eg, norepinephrine, atriopeptin, bradykinin, adenosine, nitric oxide). Afterload is often increased in heart failure, and therapy is often directed at decreasing it.
Increases in heart rate result in increasing myocardial oxygen consumption while decreasing time for diastole when coronary blood flow is greatest. The combination can set the stage for an imbalance in myocardial oxygen demand and supply, leading to myocardial ischemia. Cardiac failure is characterized by an increase in sympathetic tone and relative increases in heart rate; the ultimate impact is an inefficient myocardium that can result in deleterious remodeling.
Oxygen is responsible for the production of the vast majority of ATP, which fuels both contraction and relaxation of the myocardium. Calcium must rapidly be released by intracellular stores (sarcoplasmic reticulum) via calcium-induced calcium release to allow for excitation-contraction coupling, while equally rapid removal of calcium back into the sarcoplasmic reticulum is necessary for relaxation. Both processes of calcium cycling are energy dependent.
In heart failure and cardiomyopathy, inappropriate handling of calcium may result in arrhythmogenesis and may also be the most important factor that leads to both reduced force of contraction and reduced rate of relaxation (ie, reduced systolic as well as diastolic function).
Hindrance to Blood Flow
Blood flow from the heart, termed cardiac output, is via both the left and right ventricles. Cardiac output is determined by the heart rate and ventricular stroke volume. Blood flows through the systemic arterial (left ventricular) or pulmonary arterial (right ventricular) trees and is critical to satisfactory function of the heart and consequent perfusion of organs with adequate quantities of blood and the oxygen it contains. Normal cardiac output for dogs and cats is 100–200 mL/kg/min and 120 mL/kg/min, respectively.
Most (>90%) of the hindrance to blood flow is from the degree of constriction of the arterioles, termed vascular resistance; however, some interference is from the stiffness of the portion of the great arteries closest to the ventricles, termed impedance. Impedance is the sum of external factors opposing left ventricular ejection and is closely related to afterload. The ventricles eject a stroke volume into the proximal portion of the great arteries, which expand to accommodate the stroke volume; when the ventricles are relaxed, the distended great arteries recoil and keep blood moving through the arterioles into the capillaries. The aortic and pulmonic valves close and prevent the stroke volume from returning to the ventricle that ejected it.
Systemic vascular resistance is the opposing blood flow that must be overcome to push blood through the peripheral circulation and is calculated by:
(mean arterial pressure – central venous pressure)/cardiac output
Right atrial pressure can also used in place of central venous pressure to calculate systemic vascular resistance.
Pulmonary vascular resistance is similarly calculated: (mean pulmonary artery pressure - pulmonary arterial wedge pressure)/cardiac output. Left atrial pressure can be substituted for pulmonary arterial wedge pressure. Pulmonary vascular resistance is increased in cases of pulmonary vascular obstruction or pulmonary hypertension.
One of the most important features of heart failure that leads to morbidity is increased resistance of arterial, arteriolar, and venous smooth muscle because of increased angiotensin II, vasopressin, and endothelin. If the left ventricle is unable to eject a normal stroke volume or cardiac output, it is reasonable that ventricular function might be improved by decreasing vascular resistance. Decreasing afterload (arterial vasodilation) is one therapeutic goal in heart failure therapy.






