

The importance of pain management and the use of nonsteroidal anti-inflammatory drugs (NSAIDs) in animals has increased dramatically in recent decades, and use of NSAIDs in companion animals is routine practice. NSAIDs have the potential to relieve pain and inflammation without the myriad potential metabolic, hemodynamic, and immunosuppressive adverse effects associated with corticosteroids. However, all NSAIDs have the potential for other adverse effects that should be considered in overall management of the inflammatory process.
Mode of Action of Nonsteroidal Anti-inflammatory Drugs in Animals
Generally, the classification NSAID is applied to drugs that inhibit one or more steps in the metabolism of arachidonic acid (AA). Unlike corticosteroids, which inhibit numerous pathways, NSAIDs act primarily to reduce the biosynthesis of prostaglandins by inhibiting cyclooxygenase (COX). In general, NSAIDs do not inhibit the formation of 5-lipoxygenase (5-LOX) and hence leukotriene, or the formation of other inflammatory mediators.
The discovery of the two isoforms of COX (COX-1 and COX-2) has led to greater understanding of the mechanism of action and potential adverse effects of NSAIDs. COX-1, expressed in virtually all tissues of the body (eg, gut and kidney), catalyzes the formation of constitutive prostaglandins, which mediate a variety of normal physiologic effects, including hemostasis, gastrointestinal mucosal protection, and protection of the kidney from hypotensive insult. In contrast, COX-2 is activated in damaged and inflamed tissues and catalyzes the formation of inducible prostaglandin, including PGE2, associated with intensifying the inflammatory response. COX-2 is also involved in thermoregulation and the pain response to injury. Therefore, COX-2 inhibition by NSAIDs is thought to be responsible for the antipyretic, analgesic, and anti-inflammatory actions of NSAIDs.
However, concurrent inhibition of COX-1 may result in many of the adverse effects of NSAIDs, including gastric ulceration and renal toxicity. Because NSAIDs vary in their ability to inhibit each COX isoform, a drug that inhibits COX-2 at a lower concentration than that necessary to inhibit COX-1 would be considered safer. This concept has propelled the development of the “COX-2 selective” NSAIDs. Although ratios of COX-1:COX-2 inhibition (usually IC50 COX-1: IC50 COX-2) by various NSAIDs in humans and animals have been reported, caution is advised when interpreting such ratios, because they vary greatly depending on the selectivity assay used. The COX selectivity of NSAIDs also varies by species; COX selectivity ratios reported for humans cannot be directly extrapolated to other species.
In general, drugs with ratios suggesting preferential activity against COX-2 may have fewer adverse effects due to COX-1 inhibition. In dogs, favorable ratios have been reported for carprofen, meloxicam, deracoxib, firocoxib, and robenacoxib, whereas unfavorable ratios have been reported for aspirin, phenylbutazone, and vedaprofen. COX-1–sparing drugs are associated with less gastrointestinal ulceration and less platelet inhibition; however, it may be an oversimplification to assume that complete COX-2 inhibition is without risk.
Recent research has suggested that COX-2 can be induced constitutively in various organs, including the brain, spinal cord, ovaries, and kidneys. In dogs, COX-2 mRNA is present in the loop of Henle and the maculae densa and may play an important role in the protective response to hypotension. Renal expression of COX-2 varies between species, being greater in dogs compared with humans. COX-2 also appears to be important in the healing of gastrointestinal ulcers in humans, and certain COX-2–specific inhibitors delay ulcer healing experimentally. Although COX-1 plays a primary role in regulating homeostasis, it may play a more important role in inflammation than originally proposed.
NSAIDs enter the pocket of the COX enzyme, whereupon steric hindrance prevents entry of AA. Aspirin is unusual in that it irreversibly acetylates a serine residue of COX, resulting in a complete loss of COX activity. Thus, the duration of the aspirin effect depends on the turnover rate of COX; activity is lost for the life of the platelet (7–10 days) after aspirin administration, explaining the duration of aspirin’s effect on hemostasis. Unlike aspirin, most other NSAIDs (including salicylic acid, an active metabolite of aspirin) are reversible competitive COX inhibitors; their duration of inhibition is primarily determined by the elimination pharmacokinetics of the drug.
Pharmacologic Effects of Nonsteroidal Anti-inflammatory Drugs in Animals
All NSAIDs, except for acetaminophen (also named paracetamol), are antipyretic, analgesic, and anti-inflammatory. They are routinely used for the relief of pain and inflammation associated with osteoarthritis in dogs and horses and for colic, navicular disease, and laminitis in horses. The use of NSAIDs for the relief of perioperative pain in companion animals is standard practice. In general, NSAIDs provide only symptomatic relief from pain and inflammation and do not significantly alter the course of pathologic damage. As analgesics, they are generally less effective than opioids and are therefore generally indicated only against mild to moderate pain in humans. However, in veterinary medicine, NSAIDs also find use in management of severe pain, optimally in combination with an opioid.
As antipyretics, NSAIDs reduce body temperature in febrile states. Although the beneficial effects of the febrile response usually outweigh the negative effects, NSAID inhibition of PGE2 activity in the hypothalamus may provide symptomatic relief and improve appetite. In the EU, NSAIDs have been used in conjunction with antimicrobials for treatment of acute respiratory diseases in cattle. They may reduce morbidity via their antipyretic and anti-inflammatory effects and prevent development of irreversible lung lesions.
The effects of some NSAIDs on chondrocyte metabolism have been investigated. Some, including aspirin, naproxen, and ibuprofen, are considered chondrotoxic, because they inhibit the synthesis of cartilage proteoglycans. Others, including carprofen and meloxicam, may be considered chondroneutral or, depending on dose, actually stimulate the production of cartilage matrix. The potential beneficial or deleterious effects of NSAIDs on chondrocyte metabolism remain to be clarified.
A therapeutic area in which NSAID use may become important is in the treatment and prevention of cancer. Epidemiologic studies in humans show that aspirin use is associated with a significant reduction in the incidence of colon cancer. Newer evidence suggests that the therapeutic effect of NSAIDs on colon cancer is mediated by inhibition of COX-2, which may be upregulated in many premalignant and malignant neoplasms. In veterinary medicine, piroxicam has been shown to reduce the size of tumors such as transitional cell carcinoma in dogs. Specific COX-2 inhibitors may prove useful as a primary or adjunctive treatment in the management of cancer.
Administration and Pharmacokinetics of Nonsteroidal Anti-inflammatory Drugs in Animals
Most NSAIDs are weak organic acids that are well absorbed after oral administration. However, food can impair the oral absorption of some NSAIDs (eg, phenylbutazone, meclofenamate, flunixin, and robenacoxib). Several NSAIDs are available as parenteral formulations for intravenous, intramuscular, or subcutaneous administration. Some parenteral formulations are highly alkaline (eg, phenylbutazone) and may cause tissue necrosis if injected perivascularly. Once absorbed, most NSAIDs are extensively (up to 99%) bound to plasma proteins, with only a small proportion of unbound drug available to be active in the tissues. NSAIDs may also compete for binding sites with other highly protein-bound compounds, leading to some drug displacement; however, this displacement has little therapeutic consequence, because it does not affect the concentration of the free drug. Because NSAIDs are highly protein bound and extravasation of protein occurs in inflammation, NSAIDs tend to concentrate in areas of inflammation. Consequently, their duration of action typically exceeds that predicted by elimination half-life.
Most NSAIDs are biotransformed in the liver to inactive metabolites that are excreted either by the kidney via glomerular filtration and tubular secretion or by the bile. Mavacoxib is an exception, mostly being excreted unchanged in the bile. Biotransformation and elimination half-lives vary significantly by species (and in some cases by breed or strain, as is the case for some COX-2 inhibitors in Beagles), so it is not possible to safely extrapolate dosages from one species or animal to another. Some NSAIDs, including naproxen, etodolac, and meclofenamic acid, undergo extensive enterohepatic recirculation in some species, resulting in prolonged elimination half-lives.
Adverse Effects of Nonsteroidal Anti-inflammatory Drugs in Animals
All NSAIDs have the potential to induce adverse reactions, some of which can be life-threatening. Many reactions to NSAIDs are dose-related and are typically reversible with discontinuation of treatment and supportive care.
Vomiting is the most commonly observed adverse effect, with gastrointestinal ulceration the most common life-threatening adverse effect. Loss of gastrointestinal protective mechanisms results from inhibition of COX-1 constitutive prostaglandins that regulate blood flow to the gastric mucosa and stimulate bicarbonate and mucus production. This disrupts the alkaline protective barrier of the gut, allowing diffusion of gastric acid back into the mucosa, injuring cells and blood vessels, and causing gastritis and ulceration. The organic acid NSAIDs, especially aspirin, may also cause direct chemical irritation of the gastrointestinal mucosa and uncouple oxidative phosphorylation in mucosal epithelial cells.
The enterohepatic recirculation of certain NSAIDs may result in high biliary concentrations that increase ulcerogenic potential in the gut. Small intestine protein-losing enteropathy is a further manifestation of COX-1 inhibition and enterohepatic recycling. NSAID-induced gastrointestinal bleeding may be occult, leading to iron-deficiency anemia, or be more severe, resulting in vomiting, hematemesis, and melena.
In horses, right dorsal colitis (leading to protein-losing enteropathy) may occur, with accompanying signs of colic and diarrhea. Hypoalbuminemia may be evident on clinical pathologic evaluation, and there is potential for progression to ulceration and perforation. Theoretically, COX-2 selective (COX-1 sparing) NSAIDs should cause few or no gastrointestinal adverse effects. However, COX-2 selective NSAIDs have been shown to cause gastritis, erosion, ulceration, and enteropathy in humans and animals. COX-2 is also involved in protective mechanisms of the gastric mucosa; COX-2 selectivity confers relative gastrointestinal safety.
Gastrointestinal blood loss may be further complicated by impaired platelet function; NSAIDs, by inhibiting COX-1, prevent platelets from forming TXA2, a potent aggregating agent. Delayed clotting time would therefore be expected, and this occurs with aspirin but much less so with COX-2 selective NSAIDs. Aspirin irreversibly binds to platelet COX-1; however, with other NSAIDs, the binding is competitive. Clinically, at least in the dog, COX-2 selective NSAIDs do not appear to substantially delay clotting time (based on buccal mucosal bleeding time, prothrombin time, and partial thromboplastin time). Meloxicam and other NSAIDs have been licensed for preoperative administration for routine spay neuter surgeries, however caution is indicated in patients at increased risk of hemorrhage. Because TXA2 inhibition causes prolonged bleeding, evaluation of buccal mucosal bleeding time is advised in animals for which surgery is anticipated. Blood dyscrasias after longterm treatment with NSAIDs other than aspirin have been reported in cats, dogs, and horses. Acetaminophen (paracetamol) administration in cats is associated with Heinz body anemia, methemoglobinemia, hepatic failure, and death. Bone marrow dyscrasias associated with phenylbutazone administration have also been reported.
COX-2 prostaglandins mediate protective renal vasodilation during hypotension. Animals with underlying renal compromise receiving NSAIDs could experience exacerbation or decompensation of their disease. Maintaining hydration and renal perfusion is important in animals receiving NSAIDs, especially those undergoing anesthesia or surgery, and in horses with colic. COX-2 selective NSAIDs are not safer regarding potential for nephropathies, including papillary necrosis and interstitial nephritis.
NSAID administration may induce mild hepatic changes, characterized primarily by increases in liver enzymes without clinical signs or hepatic dysfunction. Rare reports of idiosyncratic reactions resulting in hepatic dysfunction or failure have been reported in humans (acetaminophen and others), dogs (acetaminophen, carprofen, etodolac), and horses (phenylbutazone). Cytopathic (hepatocellular injury, necrosis), cholestatic, and mixed histopathologic patterns of injury have been documented. NSAIDs should be used with caution in animals with preexisting hepatic disease.
Specific Nonsteroidal Anti-inflammatory Drugs
Based on structure, most NSAIDs can be divided into three broad groups: carboxylic acid and enolic acid derivatives, and the COX-2 inhibitors. Carboxylic acid subgroups include the salicylates (aspirin), propionic acids (ibuprofen, naproxen, carprofen, ketoprofen, and vedaprofen), fenemates (tolfenamic and meclofenamic acids), phenylacetic acids (acetaminophen), and aminonicotinic acids (flunixin). The main subgroups of enolic acids are the pyrazolones (phenylbutazone) and the oxicams (meloxicam, piroxicam).
The newer coxib class of selective COX-2 inhibitors has burgeoned in recent decades with a checkered track record in human medicine and a plethora of members entering the veterinary field; cimicoxib even making the transition from human into veterinary medicine. NSAIDs include deracoxib, firocoxib, robenacoxib, mavacoxib, and cimicoxib. The only COX-2 inhibitor remaining in human medicine in the US is celecoxib, after rofecoxib, veldecoxib, and lumiracoxib were withdrawn after being shown to increase the risk of heart attack and strokes in humans. In other countries, parecoxib (the prodrug of valdecoxib) and etoricoxib are also available for human use.
Although NSAIDs may differ in their relative potencies, they are all equally efficacious at the appropriate dose rates.
Grapiprant is a novel PGE2 antagonist, acting at the EP4 receptor. It is not a COX-1 or COX-2 inhibitor.
Aspirin Use in Animals
By far the most widely used NSAID in humans, aspirin is primarily used in veterinary medicine for relief of mild to moderate pain associated with musculoskeletal inflammation or osteoarthritis. The salicylic ester of acetic acid, aspirin (acetylsalicylic acid) is available in several different dosage forms, including bolus (for cattle), oral paste (for horses), oral solution (for poultry), and tablets (for dogs). Enteric-coated products used in human medicine are not recommended in dogs, because gastric retention may lead to erratic plasma exposure.
After oral administration, aspirin is rapidly absorbed from the stomach and upper small intestine. Aspirin is subjected to a large first-pass effect in the liver to yield salicylic acid, its main active metabolite. In addition, the aspirin fraction that gains access to the systemic circulation is also rapidly hydrolyzed to salicylic acid with a half-life of ~15 minutes. After oral aspirin administration, salicylic acid is considered the main active substance in the systemic circulation. Aspirin primarily inhibits COX-1, whereas salicylic acid has more balanced COX-1/COX-2 activity. In addition, aspirin may irreversibly bind to COX-1 through acetylation of a serine residue near the enzyme active site. Because of this irreversible binding, the anticoagulant activity of aspirin lasts far longer than its anti-inflammatory effect; a single aspirin dose of 20 mg/kg in a horse may prolong bleeding for 48 hours.
Depending on its route of administration, aspirin may have different pharmacologic effects. For irreversible platelet COX-1 inhibition (to treat a thromboembolic condition), aspirin administered intravenously is more efficient than aspirin administered orally because, for the same dose, aspirin exposure is greater for the intravenous route of administration.
After absorption, both aspirin and salicylate are widely distributed through most tissues and fluids and readily cross the placental barrier. Approximately 80%–90% of salicylate is bound to plasma proteins. Metabolism and elimination is via hepatic conjugation with glucuronic acid, followed by renal excretion. Cats, which lack glucuronyl transferase, metabolize salicylates slowly. In addition, salicylate metabolism is saturable and, if overexposure due to an aspirin overdose occurs, plasma salicylate elimination may follow a zero order and slower elimination kinetics. The elimination half-life of salicylic acid in cats approaches 40 hours, whereas it is ~7.5 hours in dogs.
Because aspirin is not approved for veterinary use, definitive efficacy studies have not been performed to establish effective dosages. Recommended dosages in dogs are 10–40 mg/kg, orally, every 8–12 hours. Aspirin has been used for its anticlotting effect in the treatment of laminitis in horses at a dosage of 10 mg/kg per day, PO. In cats, aspirin may be used for its antiplatelet effects in thromboembolic disease at a dosage of 10 mg/kg, PO, every 48 hours, to allow for prolonged metabolism. Adverse effects are common after aspirin administration and appear to be dosage dependent. Even at therapeutic dosages of 25 mg/kg, plain aspirin may induce mucosal erosion and ulceration in dogs. Vomiting and melena may occur at higher doses. The PGE1 analogue misoprostol may decrease gastrointestinal ulceration associated with aspirin and other NSAIDs. Aspirin overdose in any species can result in salicylate poisoning, characterized by severe acid-base abnormalities, hemorrhage, seizures, coma, and death.
Acetaminophen in Animals
Acetaminophen (paracetamol) is a para-aminophenol derivative with analgesic and antipyretic effects similar to those of aspirin, but it has weaker anti-inflammatory effects than does aspirin and other NSAIDs. The reason for this anomaly is that acetaminophen’s selective COX-2 inhibition is via enzyme reduction; the high levels of peroxides in areas of inflammation are thought to interfere with COX-2 reduction peripherally, whereas the low peroxide levels in the brain and spinal cord account for any centrally mediated analgesia. Acetaminophen does not inhibit neutrophil activation, has little ulcerogenic potential, and has no effect on platelets or bleeding time. The recommended dosage of acetaminophen in dogs is 10–15 mg/kg, PO, every 8 hours. Dose-dependent adverse effects include depression, vomiting, and methemoglobinemia. Acetaminophen has been used in horses; however, the therapeutic range for efficacy and appropriate dose rates are yet to be established. Use in cats is contraindicated because of their deficiency of glucuronyl transferase, which makes them susceptible to methemoglobinemia and centrilobular hepatic necrosis.
Acetaminophen in combination with codeine has been used in dogs. The efficacy of oral codeine is weak, because very little is converted to morphine in this species, which queries the benefit of using the acetaminophen-codeine combination. In fixed combination formulations, the ability for the veterinarian to independently make dose adjustments of each constituent drug is prevented; increasing the dose of codeine in pursuit of improved analgesia may cause acetaminophen toxicity.
Phenylbutazone in Animals

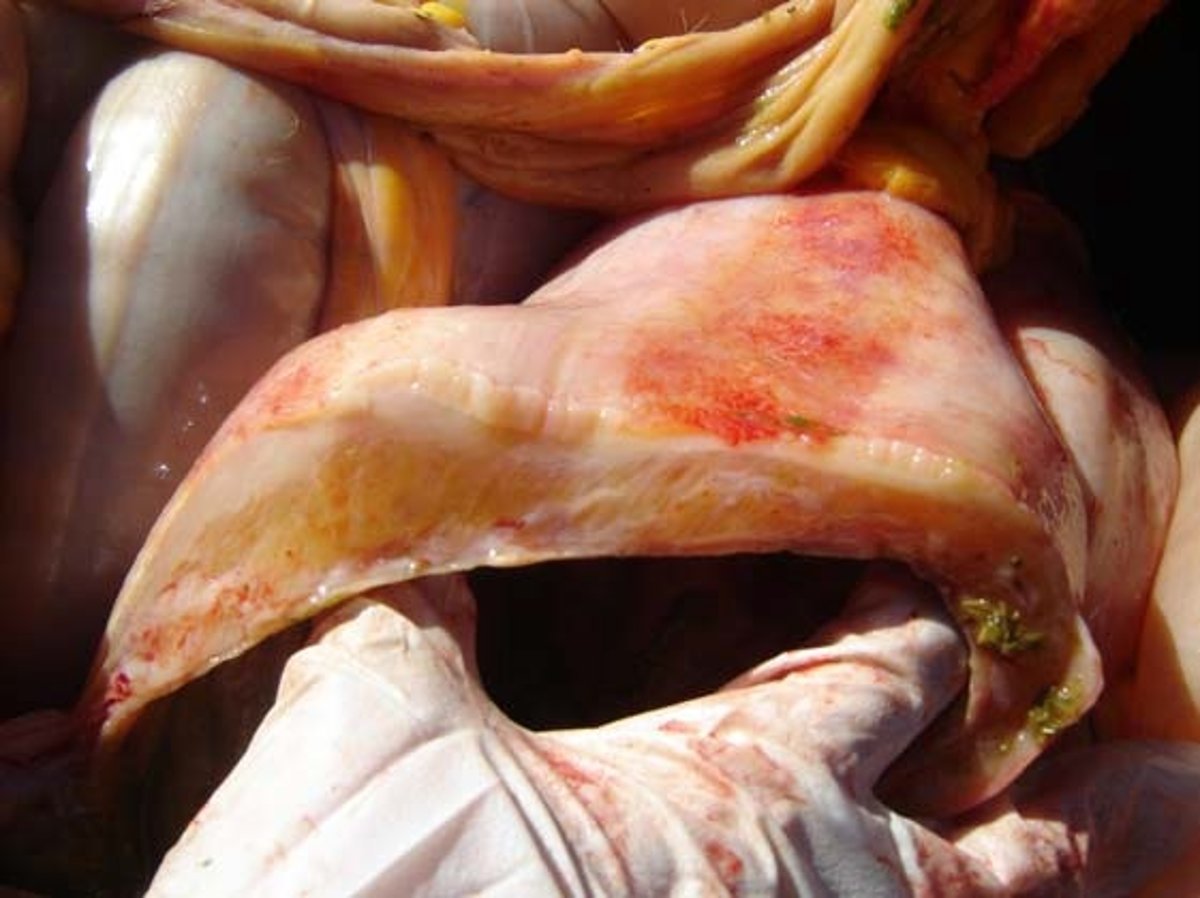
Photograph of gross pathology of right dorsal colitis in a horse caused by a course of phenylbutazone at label dosing.
Courtesy of Dr. Sharanne Raidal.
One of the earliest NSAIDs approved for use in horses and dogs, phenylbutazone (PBZ) is a pyrazolone derivative available in tablet, paste, gel, and parenteral formulations. When administered orally PBZ adsorbs to hay in the diet, to then be released during fermentation in the hindgut. Although this potentially may reduce gastrointestinal absorption and bioavailability, the clinically relevant effect is a delay in absorption. Once PBZ is absorbed, binding to plasma proteins is high (99% in horses). PBZ is metabolized by the liver to several active (oxyphenbutazone) and inactive metabolites, which are excreted in urine.
One of the major therapeutic uses of PBZ is to treat acute laminitis in horses. Laminitis is treated initially with injectable PBZ at dosages up to 8.8 mg/kg, followed by treatment at 2.2–4.4 mg/kg, PO, every 12 hours. Because the therapeutic index for PBZ is relatively narrow (PBZ exhibits zero order metabolism), the dosage should be adjusted to the minimum possible to maintain comfort and avoid toxicity. Gastrointestinal effects (eg, anorexia) and depression are the most frequent adverse effects associated with PBZ. Ulcers may develop in the mouth, stomach, cecum, and right dorsal colon. The ulcerogenic potential of PBZ in horses is greater than that of flunixin and ketoprofen. PBZ dosages of 3–7 mg/kg, PO, every 8 hours, are recommended in dogs. In dogs, PBZ has been associated with bleeding dyscrasias, hepatopathies, nephropathies, and rare cases of irreversible bone marrow suppression.
The plasma half-life of PBZ is 5–6 hours in horses and dogs. As an example of a major species difference in drug metabolism, cattle clear PBZ extremely slowly, resulting in an elimination half-life of more than 30 days. Although PBZ is not approved for use in cattle because of residue concerns (PBZ-induced blood dyscrasias in humans), extra-label use in dairy cattle prompted the FDA to prohibit its use in dairy cows 20 months of age and older.
Meclofenamic Acid in Animals
Meclofenamic acid is a fenemate (anthranilic acid) NSAID available for horses as a granular preparation and for dogs as an oral tablet. The recommended dosage in horses is 2.2 mg/kg per day for 5–7 days; the recommended dosage in dogs is 1.1 mg/kg per day for 5–7 days. In cattle, administration of meclofenamic acid results in a biphasic pattern of absorption, with an initial peak plasma concentration reached at ~30 minutes and a secondary peak 4 hours after dosing. In horses, meclofenamic acid is rapidly absorbed, but feeding before dosing may delay absorption. The onset of action is slow, requiring 2–4 days of dosing for a clinical effect. Although it is effective in the treatment of chronic laminitis, meclofenamic acid has a therapeutic index that may be lower than that of other NSAIDs.
Tolfenamic Acid in Animals
Tolfenamic acid is a fenemate NSAID, and it is a structural analogue of flunixin. It is approved for use in the EU and other countries. It is used for fever, postoperative pain, and acute and chronic inflammatory conditions in cats, dogs, cattle, and pigs.
Flunixin in Animals
Flunixin is a nonselective COX inhibitor. In the US, the nicotinic acid derivative flunixin (as the meglumine salt) is approved for use in horses as oral and parenteral formulations. A topical formulation also has been approved for use in cattle. The recommended dosage is 1.1 mg/kg per day for 5 days, PO or IV. Flunixin is rapidly absorbed after oral or intramuscular administration, and the elimination half-life is short (~2–3 hours). Elimination is primarily by renal excretion. Flunixin is effective for the treatment of musculoskeletal inflammation and visceral pain associated with colic in horses. It is also used to reduce the inflammatory-mediated hemodynamic response to endotoxin, although it is unlikely to reduce mortality associated with endotoxemic shock. The "anti-endotoxin" assertion stems from experimental studies in which horses were administered flunixin prior to endotoxin challenge, and inflammatory marker and hemodynamic changes measured thereafter; mortality was not reduced, and NSAIDs have been comprehensively shown in humans to have no impact on mortality associated with endotoxic shock.
The dosage recommended in horses is 1.1 mg/kg, every 12 hours, or 0.25 mg/kg, every 8 hours. Toxicity in horses is relatively uncommon, but gastrointestinal erosion and ulceration may develop. Flunixin has been used to treat mastitis and acute pulmonary emphysema in cattle, although it is not approved for these indications. Chronic administration of flunixin to dogs may result in severe gastrointestinal ulceration and renal damage. Flunixin is not marketed in the US for dogs, but it is approved in the EU and other countries.
Carprofen in Animals
Carprofen is an NSAID of the arylpropionic acid class available in the US in caplet and chewable tablet formulations. An injectable formulation is also available in the US and the EU. Carprofen is approved by the Food and Drug Administration to manage pain and inflammation associated with osteoarthritis and acute pain associated with soft-tissue and orthopedic surgery in dogs. The recommended dosage is 4.4 mg/kg per day or divided every 12 hours, PO. In the EU and other countries, carprofen is also registered for use in horses and cattle and for short-term treatment in cats. In dogs, oral bioavailability is high (90%), and plasma concentrations peak ~2–3 hours after dosing. The elimination half-life is ~8 hours. As with other NSAIDs, carprofen is highly (99%) protein bound. Elimination is via hepatic biotransformation, with excretion of the resulting metabolites in feces and urine. Some enterohepatic recycling occurs.
The exact mechanism of action of carprofen is unclear. Although it has greater selectivity for COX-2 over COX-1, carprofen is considered a weak COX inhibitor. In vitro assays with canine cell lines indicate that it is 129-fold more selective for COX-2, whereas in vitro assays with canine whole blood indicate that it is 7- to 17-fold more selective for COX-2. Equine whole blood assays indicate that it is 1.6-fold more selective for COX-2, and feline whole blood assays indicate it is >5.5-fold more selective for COX-2. Other mechanisms of action, including inhibition of PA2, may be responsible for its anti-inflammatory effects.
Carprofen has been used extensively in dogs since its introduction, and adverse events have been comparable to those of other NSAIDs (ie, ~2 events/1,000 dogs treated). Approximately one-fourth of the adverse reactions reported were gastrointestinal signs, including vomiting, diarrhea, and gastrointestinal ulceration. Renal and hepatic adverse effects are rare, as with other NSAIDs. Potentially serious idiosyncratic hepatopathies, characterized by acute hepatic necrosis, have been reported in some dogs. Approximately one-third of the dogs developing hepatopathies while receiving carprofen were Labrador Retrievers, although a true breed predisposition has not been established. As with any NSAID treatment, clinical laboratory monitoring for hepatic damage is advised, especially in geriatric animals that may be predisposed to more serious complications.
Ketoprofen in Animals
Ketoprofen is another propionic acid derivative available in the US and other countries as a 10% injectable solution for horses, and in the EU and Canada as tablets and a 1% injectable solution for dogs and cats. Ketoprofen is recommended for acute pain (up to 5 days) in both dogs and cats. In horses, it is used for pain and inflammation associated with osteoarthritis and for visceral pain associated with colic. The recommended dosage is 1 mg/kg per day for up to 5 days, IV or PO in dogs and cats; 2.2 mg/kg per day for up to 5 days, IV, in horses; and 3 mg/kg per day for 1–3 days, IV or IM, in cattle. Ketoprofen is a potent inhibitor of COX and bradykinin and may also inhibit some lipoxygenases. Its efficacy is comparable to that of opioids in the management of pain after orthopedic and soft-tissue surgery in dogs. After oral administration, ketoprofen is rapidly absorbed and has a terminal half-life in cats and dogs of 2–3 hours. As with other NSAIDs, ketoprofen is metabolized in the liver to inactive metabolites that are eliminated by renal excretion. Adverse effects, including gastrointestinal upset, are similar to those of other NSAIDs. Other adverse effects, including hepatopathies and renal disease, have been reported in animals. Because of potential antiplatelet effects, care should be exercised when using ketoprofen perioperatively.
Etodolac in Animals
The pyranocarboxylic acid etodolac is approved for use in dogs in the US. The elimination half-life is ~8–12 hours, allowing dosing at 10–15 mg/kg per day, PO. Extensive enterohepatic recirculation has been reported in dogs, followed by elimination of etodolac and its metabolites in the liver and feces. In in vitro studies, etodolac was more selective in inhibiting COX-2 than COX-1, although in vitro canine whole blood assays have also shown it to be nonselective. Etodolac has been shown to inhibit macrophage chemotaxis and has demonstrated efficacy for the treatment of lameness associated with hip dysplasia. Although the risk of gastrointestinal ulceration is low at therapeutic doses, administration of three times the label dosage resulted in gastrointestinal ulceration, vomiting, and weight loss in toxicity studies. gastrointestinal, hepatic, and renal adverse reactions have been reported after administration of etodolac, similar to those of other NSAIDs.
Vedaprofen in Animals
The arylpropionic acid derivative vedaprofen is available in the EU in a gel formulation for horses and dogs and in an injectable formulation for horses. Vedaprofen is indicated for the treatment of pain and inflammation associated with musculoskeletal disorders in dogs (0.5 mg/kg per day) and horses (1 mg/kg, every 12 hours) and for the treatment of pain associated with colic in horses (2 mg/kg, IV, as a single injection). After oral administration, vedaprofen is rapidly absorbed. Bioavailability is generally high but may be reduced if the drug is administered with food. The terminal half-life is 10–13 hours in dogs and 6–8 hours in horses. Vedaprofen undergoes extensive biotransformation to hydroxylated metabolites, which are excreted in urine and feces.
Meloxicam in Animals
Meloxicam is an oxicam NSAID available in a variety of formulations for various routes of administration, including an injectable solution, oral suspension, transmucosal gel, and tablet. The oral suspensions are commonly used in small animals because of ease of administration and the ability to deliver the actual calculated dose. Meloxicam is approved for human use in the US and Canada and for use in dogs in the US. In the EU and other countries, it is approved for use in dogs, cats, cattle, pigs, and horses, and, in Australia, sheep.
A potent inhibitor of prostaglandin synthesis, meloxicam is used for the treatment of acute and chronic inflammation associated with musculoskeletal disease and for the management of postoperative pain. In dogs, a one-time loading dose of 0.2 mg/kg, PO, is recommended, followed by 0.1 mg/kg per day, PO. Once a therapeutic effect occurs, the dosage can be titrated to the lowest possible dose. COX-1:COX-2 ratios reported for meloxicam suggest the drug is COX-2 selective, with in vitro canine whole blood assays indicating it is 2.7- to 10-fold more selective for COX-2. Once absorbed, meloxicam is highly protein bound (97%) and has a relatively long elimination half-life (>12 hours). In humans and horses, there are no phase 2 metabolites. Gastrointestinal safety appears to be greater for meloxicam than for nonselective NSAIDs, and meloxicam has been shown to be chondroneutral in rodent studies.
In cats, meloxicam undergoes oxidative metabolism, not glucuronidation, with 80% of the drug excreted in feces. In the US, meloxicam is not approved for use in cats beyond a single injection (and a product black box warning issued to this effect), whereas in the EU and other countries the oral suspension is registered for multiple administration in cats, with a label maintenance dose of 0.05 mg/kg. There is considerable evidence that low-dose meloxicam (0.02 mg/kg) is relatively well tolerated and safe for chronic administration in cats, including those with chronic kidney disease (although staging and thorough patient evaluation before this use is prudent).
Deracoxib in Animals
Deracoxib, the first NSAID of the coxib class approved for use in dogs, is available in a beef-flavored chewable tablet formulation in the US. Deracoxib has been shown to inhibit COX-2–mediated PGE2 production. COX-1:COX-2 ratios reported for deracoxib in in vitro cloned canine cell assays indicate it is 1,275-fold more selective for COX-2, whereas in vitro canine whole blood assays indicate it is 12- to 37-fold selective for COX-2. Deracoxib is indicated for the control of postoperative pain and inflammation associated with orthopedic surgery at a dosage of 3–4 mg/kg per day for up to 7 days, PO, and for the control of pain and inflammation associated with osteoarthritis at a dosage of 1–2 mg/kg per day, PO. Once absorbed, protein binding is >90%, and the elimination half-life is 3 hours.
Firocoxib in Animals
Firocoxib is a coxib-class NSAID approved in the US and EU for the control of pain and inflammation associated with osteoarthritis and for the control of postoperative pain and inflammation associated with soft-tissue and orthopedic surgery in dogs. In Canada, Australia, and New Zealand it is approved for use in osteoarthritis and soft-tissue and orthopedic surgery. It is available in a chewable tablet formulation. After oral administration, firocoxib is rapidly absorbed and then eliminated by hepatic metabolism and fecal excretion. The elimination half-life is ~8 hours, allowing dosing at 5 mg/kg per day, PO. COX-1:COX-2 ratios from in vitro canine whole blood assays indicate it is 384-fold more selective for COX-2. As with other NSAIDs, protein binding is high, at ~96%. gastrointestinal safety appears to be greater than that of nonspecific NSAIDs.
Firocoxib is also approved for use in horses for the treatment of osteoarthritis, for a course of up to 14 days duration. In addition to an injectable formulation, tablets and an oral paste are also available.
Robenacoxib in Animals
Robenacoxib is a coxib-class highly selective COX-2 inhibitor, structurally related to the human NSAIDs diclofenac and lumiracoxib. Robenacoxib is used for the control of pain and inflammation associated with osteoarthritis, orthopedic and soft-tissue surgery in dogs (approved in EU), and for musculoskeletal disorders and soft-tissue surgeries in cats (approved in the US and EU). Dosage is 2 mg/kg, PO, initially and then 1–2 mg/kg per day thereafter (for up to 6 days in cats). COX-1:COX-2 ratios from in vitro canine whole blood assays indicate it is 128-fold more selective for COX-2. As with other NSAIDs, protein binding is high, at ~98%. Gastrointestinal safety appears to be greater than that of nonselective NSAIDs. The elimination half-life is 1 hours after oral administration. Administration with food decreases bioavailability of robenacoxib.
Mavacoxib in Animals
Mavacoxib is a coxib-class COX-2 inhibitor approved in the EU and Australia for the control of pain and inflammation associated with degenerative joint disease in dogs. Mavacoxib is structurally related to the human NSAID celecoxib; however, substitution of a methyl group with a single fluorine atom has conferred great resistance to metabolism, resulting in an elimination half-life of 17 days in young Beagle dogs. In field trials conducted in aged dogs with osteoarthritis, the half-life was found to be even longer at 44 days, and in these older dogs, approximately 1 in 20 exhibited a half-life of >80 days.
These population pharmacokinetic studies in target patients were used to optimize the dose regimen. The long half-life means mavacoxib has a unique dose regimen: the initial dose is 2 mg/kg, PO, repeated 14 days later; thereafter, the dosing interval is 1 month, with the total course not exceeding seven doses (6.5 months). Food significantly increases bioavailability. COX-1:COX-2 ratios from in vitro canine whole blood assays indicate mavacoxib is 128-fold more selective for COX-2. As with other NSAIDs, protein binding is high, at ~98%.
Unlike other NSAIDs, mavacoxib is ultimately eliminated unchanged in the feces after biliary secretion of the parent molecule. Mavacoxib differs further from most other NSAIDs; it is a weak base (pKa of 9.57) and is highly lipid soluble; thus, it has a relatively large volume of distribution (1.6 L/kg).
Grapiprant in Animals
Grapiprant is a new NSAID, available in tablet form, with a novel mode of action. It is a PGE2 antagonist, acting at the EP4 receptor subtype. It is not a COX-1 or COX-2 inhibitor. At higher than label dosages, gastrointestinal adverse effects may occur.
Dipyrone in Animals
Dipyrone (metamizole) is an atypical NSAID with weak activity against COX-1 and COX-2. Although the mechanism(s) of actions have not been entirely elucidated, central inhibition of COX-3 appears responsible for analgesia. Dipyrone has been used in humans for nearly a century; however, risk of blood dyscrasias, in particular agranulocytosis, has caused its withdrawal in many countries, including the US. Dipyrone is effectively a prodrug, being rapidly hydrolyzed to the active metabolite 4-methylaminoantipyrine. Dipyrone injection is registered for use in horses, either alone or in combination with the anticholinergic hyoscine-N-butylbromide.
Other NSAIDs Used in Animals
A large number of prescription and nonprescription NSAIDs are available for human use. However, because of species differences in metabolism, efficacy, and toxicity, many are not recommended for use in animals. For example, in dogs, indomethacin is highly toxic to the GI tract and may result in severe ulceration, hematemesis, and melena at therapeutic doses. Piroxicam undergoes extensive enterohepatic recycling in dogs, resulting in a prolonged plasma half-life. Gastrointestinal ulceration and bleeding and renal papillary necrosis have occurred in dogs receiving piroxicam at dosages of 0.3–1 mg/kg per day.
Ibuprofen is an arylpropionic acid derivative used in dogs as an anti-inflammatory agent. However, dogs are much more sensitive to the development of gastrointestinal adverse effects from ibuprofen administration than are humans. At therapeutic doses, adverse effects seen in dogs include vomiting, diarrhea, gastrointestinal bleeding, and renal infection. Ibuprofen is not recommended for use in dogs or cats.
Naproxen has been used in horses at a dosage of 5–10 mg/kg, once to twice daily. Bioavailability is lower (~50%) for naproxen than for other NSAIDs, and the elimination half-life is ~5 hours in horses. In dogs, the elimination half-life of naproxen is 35–74 hours, presumably because of extensive enterohepatic recirculation. The pharmacokinetics in dogs also appear to be breed dependent. Because of the prolonged half-life of naproxen, dogs are extremely sensitive to its adverse effects.
Coxib class drugs, including celecoxib and valdecoxib, developed for use in human medicine are COX-2 selective. In clinical studies, the incidence of gastrointestinal ulceration in patients receiving valdecoxib or celecoxib was significantly less than that of those receiving naproxen. The use of these drugs in animals has yet to be fully investigated. One pharmacokinetic study with celecoxib in Beagles demonstrated variability in drug elimination between dogs. In that study, one subgroup of Beagles metabolized celecoxib much more rapidly than the other, with elimination half-lives of ~2 and 18 hours, respectively. Until further data are available regarding the pharmacokinetics and safety of these drugs in animals, their use in veterinary medicine is not recommended.
For More Information
Also see pet health content regarding drugs used to treat inflammation.





